DEAR FRIENDS GOOD NEWS PLEASE READ
http://ushealthtimes.com/finally-myster ... sclerosis/
regards
seeva
Finally Mystery Solved: Researchers Find The Cause of MS

Re: Finally Mystery Solved: Researchers Find The Cause of MS
Rab32 connects ER stress to mitochondrial defects in multiple sclerosis.
J Neuroinflammation. 2017 Jan 23;14(1):19.
BACKGROUND: Endoplasmic reticulum (ER) stress is a hallmark of neurodegenerative diseases such as multiple sclerosis (MS). However, this physiological mechanism has multiple manifestations that range from impaired clearance of unfolded proteins to altered mitochondrial dynamics and apoptosis. While connections between the triggering of the unfolded protein response (UPR) and downstream mitochondrial dysfunction are poorly understood, the membranous contacts between the ER and mitochondria, called the mitochondria-associated membrane (MAM), could provide a functional link between these two mechanisms. Therefore, we investigated whether the guanosine triphosphatase (GTPase) Rab32, a known regulator of the MAM, mitochondrial dynamics, and apoptosis, could be associated with ER stress as well as mitochondrial dysfunction.
METHODS: We assessed Rab32 expression in MS patient and experimental autoimmune encephalomyelitis (EAE) tissue, via observation of mitochondria in primary neurons and via monitoring of survival of neuronal cells upon increased Rab32 expression.
RESULTS: We found that the induction of Rab32 and other MAM proteins correlates with ER stress proteins in MS brain, as well as in EAE, and occurs in multiple central nervous system (CNS) cell types. We identify Rab32, known to increase in response to acute brain inflammation, as a novel unfolded protein response (UPR) target. High Rab32 expression shortens neurite length, alters mitochondria morphology, and accelerates apoptosis/necroptosis of human primary neurons and cell lines.
CONCLUSIONS: ER stress is strongly associated with Rab32 upregulation in the progression of MS, leading to mitochondrial dysfunction and neuronal death.
Free full text
J Neuroinflammation. 2017 Jan 23;14(1):19.
BACKGROUND: Endoplasmic reticulum (ER) stress is a hallmark of neurodegenerative diseases such as multiple sclerosis (MS). However, this physiological mechanism has multiple manifestations that range from impaired clearance of unfolded proteins to altered mitochondrial dynamics and apoptosis. While connections between the triggering of the unfolded protein response (UPR) and downstream mitochondrial dysfunction are poorly understood, the membranous contacts between the ER and mitochondria, called the mitochondria-associated membrane (MAM), could provide a functional link between these two mechanisms. Therefore, we investigated whether the guanosine triphosphatase (GTPase) Rab32, a known regulator of the MAM, mitochondrial dynamics, and apoptosis, could be associated with ER stress as well as mitochondrial dysfunction.
METHODS: We assessed Rab32 expression in MS patient and experimental autoimmune encephalomyelitis (EAE) tissue, via observation of mitochondria in primary neurons and via monitoring of survival of neuronal cells upon increased Rab32 expression.
RESULTS: We found that the induction of Rab32 and other MAM proteins correlates with ER stress proteins in MS brain, as well as in EAE, and occurs in multiple central nervous system (CNS) cell types. We identify Rab32, known to increase in response to acute brain inflammation, as a novel unfolded protein response (UPR) target. High Rab32 expression shortens neurite length, alters mitochondria morphology, and accelerates apoptosis/necroptosis of human primary neurons and cell lines.
CONCLUSIONS: ER stress is strongly associated with Rab32 upregulation in the progression of MS, leading to mitochondrial dysfunction and neuronal death.
Free full text
Re: Finally Mystery Solved: Researchers Find The Cause of MS
See also...
Expression of a T39N mutant Rab32 protein arrests mitochondria movement within neurites of differentiated SH-SY5Y cells
Small GTPases. 2018 Jan 7:1-4.
We have shown that multiple sclerosis (MS) and endoplasmic reticulum (ER) stress induce Rab32, an ER/mitochondria-localized small GTPase. High levels of both dominant-active (Q85L) or dominant-inactive (T39N) Rab32 are toxic to neurons. While Rab32Q85L interacts with its effector Drp1 to promote mitochondria fission, it is unclear how Rab32T39N could result as toxic to neurons. Given the perinuclear clustering of mitochondria observed upon transfection of inactive Rab32, we hypothesized Rab32T39N could stall mitochondria within neurites. The movement of mitochondria depends on kinesin-binding Miro proteins. High cytosolic [Ca2+] is bound by an EF hand motif within Miro proteins, resulting in mitochondrial arrest. Consistent with increased cytosolic [Ca2+], expression of Rab32T39N arrests mitochondria movement within neurites.
Expression of a T39N mutant Rab32 protein arrests mitochondria movement within neurites of differentiated SH-SY5Y cells
Small GTPases. 2018 Jan 7:1-4.
We have shown that multiple sclerosis (MS) and endoplasmic reticulum (ER) stress induce Rab32, an ER/mitochondria-localized small GTPase. High levels of both dominant-active (Q85L) or dominant-inactive (T39N) Rab32 are toxic to neurons. While Rab32Q85L interacts with its effector Drp1 to promote mitochondria fission, it is unclear how Rab32T39N could result as toxic to neurons. Given the perinuclear clustering of mitochondria observed upon transfection of inactive Rab32, we hypothesized Rab32T39N could stall mitochondria within neurites. The movement of mitochondria depends on kinesin-binding Miro proteins. High cytosolic [Ca2+] is bound by an EF hand motif within Miro proteins, resulting in mitochondrial arrest. Consistent with increased cytosolic [Ca2+], expression of Rab32T39N arrests mitochondria movement within neurites.

Re: Finally Mystery Solved: Researchers Find The Cause of MS
Zinc depletion activates the endoplasmic reticulum-stress sensor Ire1 via pleiotropic mechanisms.
https://www.ncbi.nlm.nih.gov/pubmed/23748779
Although zinc deficiency evokes the endoplasmic reticulum (ER)-stress response, the activating mechanism remains obscure. Here we show that in yeast cells, the ER-stress sensor Ire1 was activated upon zinc deficiency. ER-stressing stimuli activating Ire1 are known to include ER accumulation of unfolded proteins and membrane-lipid aberrancy. According to the findings presented here, zinc deficiency causes both types of abnormality.
Hepatic ZIP14-mediated zinc transport is required for adaptation to endoplasmic reticulum stress.
https://www.ncbi.nlm.nih.gov/pubmed/28673968
Extensive endoplasmic reticulum (ER) stress damages the liver, causing apoptosis and steatosis despite the activation of the unfolded protein response (UPR). Restriction of zinc from cells can induce ER stress, indicating that zinc is essential to maintain normal ER function...
https://www.ncbi.nlm.nih.gov/pubmed/23748779
Although zinc deficiency evokes the endoplasmic reticulum (ER)-stress response, the activating mechanism remains obscure. Here we show that in yeast cells, the ER-stress sensor Ire1 was activated upon zinc deficiency. ER-stressing stimuli activating Ire1 are known to include ER accumulation of unfolded proteins and membrane-lipid aberrancy. According to the findings presented here, zinc deficiency causes both types of abnormality.
Hepatic ZIP14-mediated zinc transport is required for adaptation to endoplasmic reticulum stress.
https://www.ncbi.nlm.nih.gov/pubmed/28673968
Extensive endoplasmic reticulum (ER) stress damages the liver, causing apoptosis and steatosis despite the activation of the unfolded protein response (UPR). Restriction of zinc from cells can induce ER stress, indicating that zinc is essential to maintain normal ER function...
active members shape site content. if there is a problem, speak up!
use the report button to flag problematic post content to volunteer moderators' attention.
use the report button to flag problematic post content to volunteer moderators' attention.
Re: Finally Mystery Solved: Researchers Find The Cause of MS
Thank You.
Now, what should we do
Now, what should we do
Re: Finally Mystery Solved: Researchers Find The Cause of MS
Let's hope they are finally right!!!
Summary:
Using human brain tissue samples , they found that a protein called Rab32 is present in large quantities in the brains of people with MS, but is virtually absent in healthy brain cells.
Where Rab32 is present, the team discovered that a part of the cell that stores calcium (endoplasmic reticulum or ER) gets too close to the mitochondria. The resulting miscommunication with the calcium supply triggers the mitochondria to misbehave, ultimately causing toxicity for brain cells people with Multiple Sclerosis.
By the way, this was already reported here:
http://www.thisisms.com/forum/ms-etiolo ... 29044.html
Summary:
Using human brain tissue samples , they found that a protein called Rab32 is present in large quantities in the brains of people with MS, but is virtually absent in healthy brain cells.
Where Rab32 is present, the team discovered that a part of the cell that stores calcium (endoplasmic reticulum or ER) gets too close to the mitochondria. The resulting miscommunication with the calcium supply triggers the mitochondria to misbehave, ultimately causing toxicity for brain cells people with Multiple Sclerosis.
By the way, this was already reported here:
http://www.thisisms.com/forum/ms-etiolo ... 29044.html
Re: Finally Mystery Solved: Researchers Find The Cause of MS
This concept is not 'new' as info on it has been available for at least a year and it will likely take years to evaluate properly. Without looking very hard, I was able to find basic info on this concept dating back to 2016
Is this protein the cause or result of MS? It would ultimately be either and in time we will find out...
Is this protein the cause or result of MS? It would ultimately be either and in time we will find out...
Re: Finally Mystery Solved: Researchers Find The Cause of MS
The Rab family of proteins are GTPases that help regulate many steps of membrane trafficking, including vesicle formation, vesicle movement along actin and tubulin networks, and membrane fusion.frodo wrote:Where Rab32 is present, the team discovered that a part of the cell that stores calcium (endoplasmic reticulum or ER) gets too close to the mitochondria. The resulting miscommunication with the calcium supply triggers the mitochondria to misbehave, ultimately causing toxicity for brain cells people with Multiple Sclerosis.
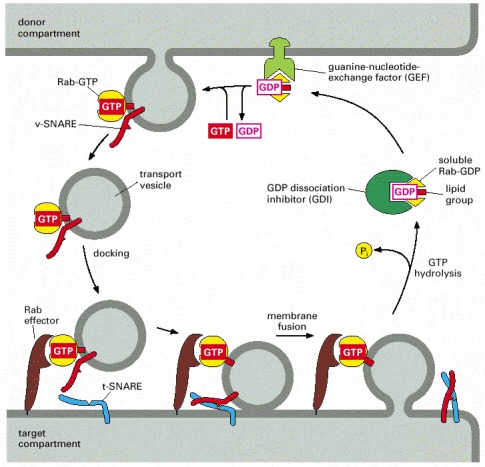
Although it's not entirely clear from this paper (though I'm still working on it), it may be the case that Rab32 allows the mitochondria to fuse with the ER thereby facilitating transfer of Ca2+.
The discussion section states that upregulation of Rab32 is downstream of ER stress.
Discussion
In our study, we report that Rab32 serves as a novel marker of neurodegeneration in MS lesions, consistent with its previously detected induction in response to proinflammatory lipopolysaccharide (LPS) [24]. Interestingly, we found that Rab32 correlated with the inflammatory status of the tissues. In contrast to healthy tissue, which showed low levels of Rab32 as reported previously [34, 35], Rab32 was highly expressed in active lesions of both human MS patients and EAE mice; while not as high, expression of Rab32 was still elevated in chronic lesions. In terms of cell types, we have detected high amounts of Rab32 in neurons and microglial/macrophage cells.
Our investigation into a transcriptional regulation of Rab32 expression showed that this gene responds to ER stress. Since ER stress is well known to trigger inflammation and mitochondrial dysfunction, our observation that ER stress leads to Rab32 induction and subsequently alters mitochondrial dynamic as well as neuronal apoptosis induction identifies Rab32 as a protein of
critical interest to MS research. Results presented in this study demonstrate that an increase of Rab32 in the inflamed brain directly promotes neuronal cell death from a combination of apoptosis and necroptosis. Interestingly, the putative role of Rab32 as an autophagy promoter [38] is not tied to this pro-death function of Rab32. While wild-type and active Rab32Q85L showed effects on mitochondrial morphology and neurite outgrowth, inactive Rab32T39N also compromised the survival of primary neurons as well as SH-SY5Y cells (Figs. 5 and 6), suggesting the mere upregulation of Rab32 is detrimental to neuronal function, potentially due to shared functions of active and inactive Rab32. Moreover, our results reinforce the role of ER stress as an upstream trigger of inflammation, which is one of the main pathology drivers in the MS context. Interestingly, the inhibition of the UPR can improve myelination of some disease models [39] and also due to shared functions of active and inactive Rab32. Moreover, our results reinforce the role of ER stress as an upstream trigger of inflammation, which is one of the main pathology drivers in the MS context. Interestingly, the inhibition of the UPR can improve myelination of some disease models [39] and also plays a critical role in the most promising approaches to treat neurodegeneration [40].
Rab32 is induced in parallel with known mediators or regulators of the MAM, namely, Grp75, PACS-2, Mitofusin 2, and Drp1 (Fig. 1). In contrast to Rab32, however, these MAM modulatory proteins were only induced in active, but not in chronic lesions. Nevertheless, this suggests that MAM functions, including the exchange of Ca2+ between the two organelles and mitochondrial dynamics, actively control neuronal decay within active MS lesions but reach a new equilibrium in chronic lesions.
-
- Similar Topics
- Replies
- Views
- Last post